

 ホーム
ホーム 最新情報
最新情報 機構紹介
機構紹介 国際交流
国際交流 サイトについて
サイトについて本ガイドラインは、「医薬品査察管理弁法(試行)」に基づき、医薬品監督管理部門によるファーマコビジランス査察の実施を指導し、医薬品販売許可保有者(以下、保有者)に対してファーマコビジランスの主体的責任を果たすよう促すために策定されたものである。
本ガイドラインは、省レベル以上の医薬品監督管理部門による、保有者自身が実施し、委託されたファーマコビジランス活動に対して行った査察業務に適用する。医薬品臨床試験の実施を承認された医薬品登録申請者に対してファーマコビジランス査察を実施する場合、医薬品の安全性特性及び臨床試験安全性情報報告並びにリスク評価と組み合わせて、臨床試験期間または販売承認前にファーマコビジランス査察を開始するものとし、具体的な実施は、本ガイドラインを参照することができる。
関連しる査察業務の組織と実施、並びに査察機関と人員、査察手順、通常査察、追加査察、査察と監査の接続、地域横断査察の協力、査察結果の処理などの関連業務は、「『医薬品査察管理弁法(試行)』の印刷・発行に関する国家医薬品監督管理局の通知」(国家医薬品監督管理〔2021〕31号)などの関連要求に従って実行されるものとする。
一、通常査察の重要な注意事項
(一)医薬品の特徴
1.医薬品の安全性特性。
2.医薬品の副作用監視データ及び副作用集中的事象の発生状況。
3.販売量が多い、または代替品が限られている医薬品。
4.販売承認時に安全性条件付き医薬品。
5.革新的な医薬品、改良型新薬、児童・妊産婦などの特殊な集団向けの医薬品。
6.社会的関心の高い医薬品。
(二)保有者の特徴
7.品種数が多く、販売量が多い保有者。
8.ファーマコビジランス査察を受けていない保有者。
9.中国国内で初めて医薬品登録証を取得した保有者。
10.企業の合併・買収、組織構造の変更等により、ファーマコビジランスシステムに重大な変更が生じるか、ファーマコビジランスの組織構造に重大な影響を与える保有者。
11.製造を委託された保有者。
12.ファーマコビジランス活動の実施を委託された保有者。
(三)その他の状況
13.以前のファーマコビジランス査察またはその他の査察の実施状況。
14.医薬品監督管理部門が査察の実施に必要の実施に必要と判断した場合。
二、追加査察の重要な注意事項
(一)副作用の疑いのある情報の報告遅延、隠蔽、漏れ、および報告の質が悪い場合。
(二)医薬品副作用監視により、安全性リスクが存在する可能性が示唆された場合。
(三)関連するリスクを適時に発見、評価、管理、または伝達することを怠る場合。
(四)製造、販売、使用を一時停止および製品のリコールを実施した後、規定に従って医薬品監督管理部門に報告しなかった場合。
(五)規定または医薬品監督管理部門の要求に従って、市販後安全性試験を実施せず、ファーマコビジランス計画を作成・実施せず、かつ理由を説明しなかった場合。
(六)医薬品監督管理部門が要求するファーマコビジランス関連情報の提供を怠る、または不適合な材料を提供した場合。
(七)是正措置の実施の遅れ、または実施が不十分な場合。
(八)その他追加査察が必要な状況。
三、査察方法
査察方法には、立入査察と遠隔査察が含まれる。立入査察とは、保有者がファーマコビジランス関連活動を行う現場で査察人員が実施する査察を指す。遠隔査察とは、ビデオ、電話などを用いて実施する査察を指す。
査察チームは、必要に応じて立入査察および/または遠隔査察を実施し、査察に必要な関連資料を指定された期限内に提出するよう保有者に要求することができる。
四、査察場所
検査場所は、主に保有者が重要なファーマコビジランス活動を実施している場所を中心に、必要に応じてファーマコビジランス活動を受託している場所への拡大査察を行う場合がある。
五、欠陥リスクレベル
ファーマコビジランス査察で発見された欠陥は、重大な欠陥、主要な欠陥、一般的な欠陥に分けられ、それらのリスクレベルは順番に低下する。前回の査察で発見された欠陥が繰り返し発生する場合は、リスクレベルを上げることができる。査察項目は合計100項目で、そのうち12項目が重大な欠陥(**)、40項目が主要な欠陥(*)、残りの48項目が一般的な欠陥と判定できる(詳細は別添を参照)。
六、評価基準
査察結論と総合評価結論は、合格、大まかに合格、不合格に分けられる。査察チーム及び派遣された査察機関は、実際の査察状況に基づき、以下の評価基準を参照し、査察結論と総合評価結論を出すことができる。
(一)重大な欠陥項目と主要な欠陥項目が発見されず、一般的な欠陥項目が0~9項目以内、合格と評価できる。
(二)次のいずれかの条件に該当する場合、不合格と評価できる。
1.重大な欠陥項目が1つ以上。
2.重大な欠陥項目がなく、かつ主要な欠陥項目は10項以上。
2.重大な欠陥項目がなく、主要な欠陥項目は0~9項、欠陥の合計が25項以上。
(三)それ以外の場合は、大まかに合格と評価できる。
別添:ファーマコビジランス査察の重点
別添
ファーマコビジランス査察の重点
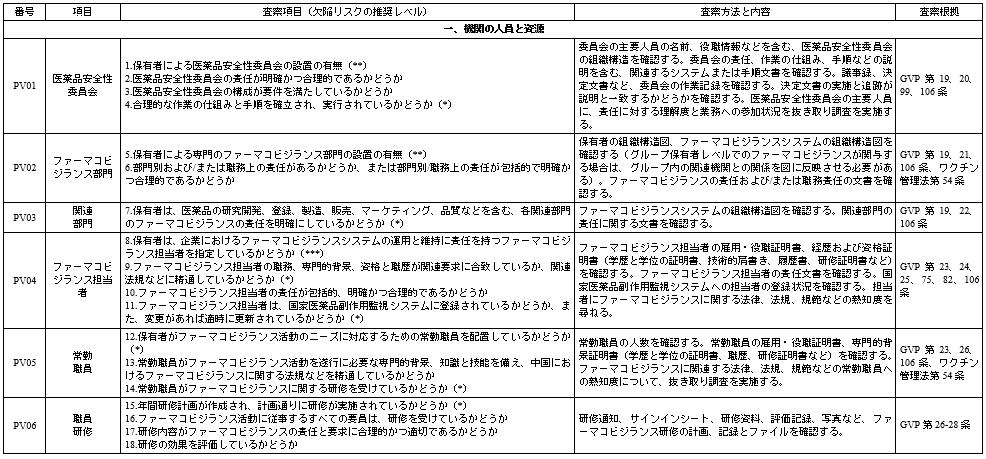
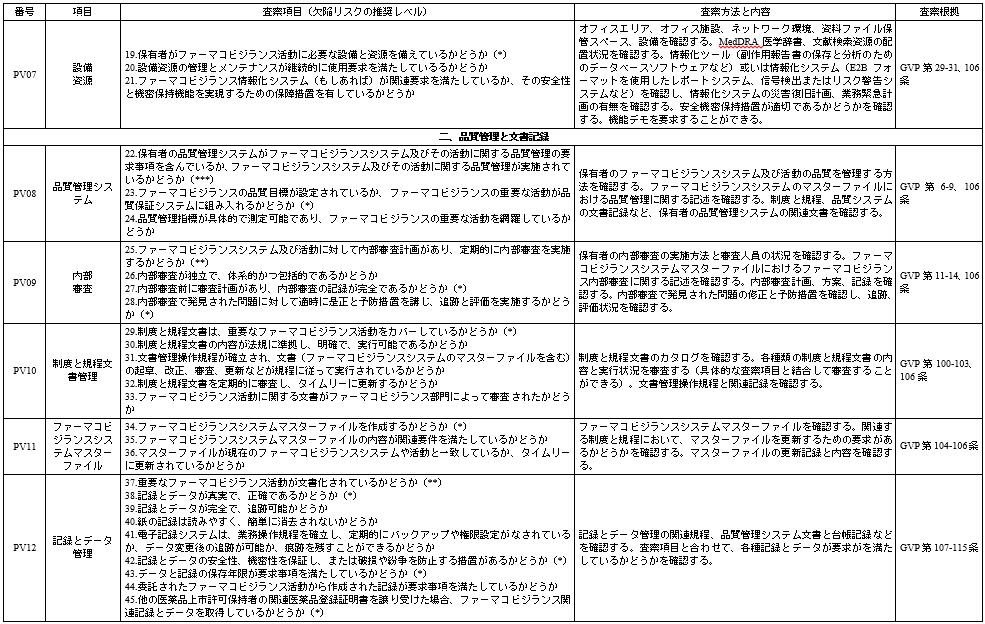
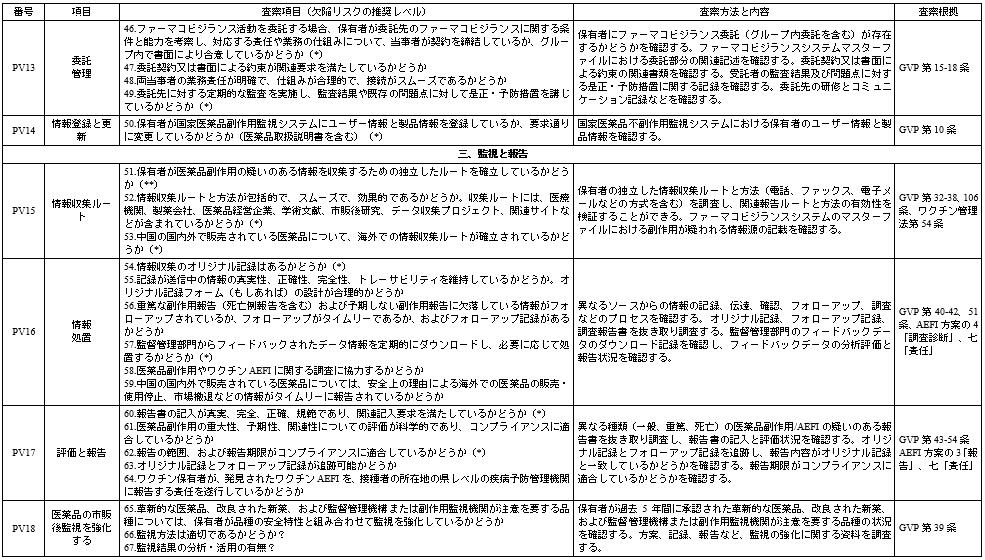
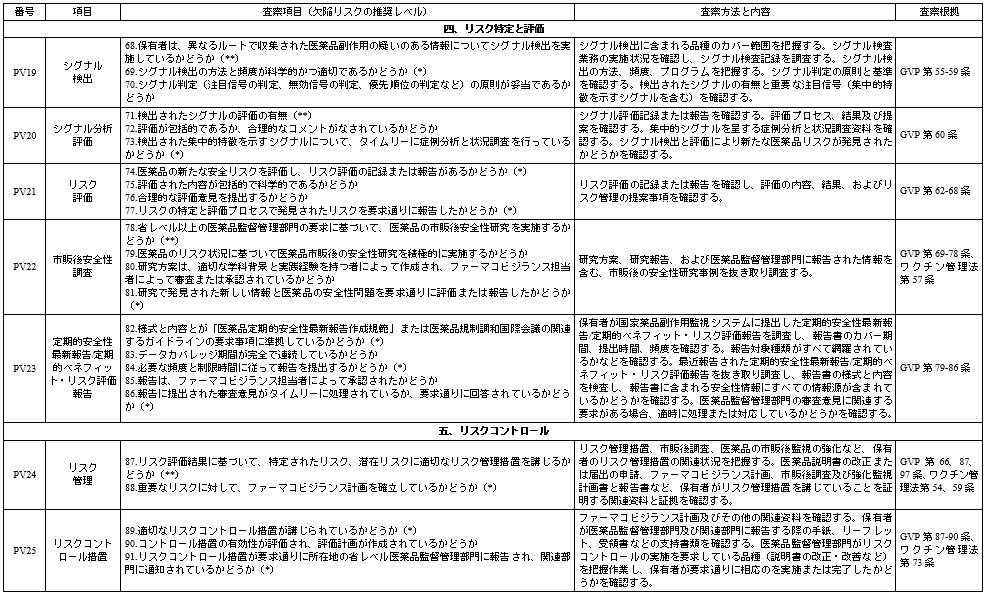

注:1. 保有者に提供を求める関連資料は、概ね3年以内のもの、あるいは前回の査察から今回の査察までの期間に形成された資料である。
2. 本表で、GVPは「ファーマコビジランス品質管理規範」、AEFI方案は「全国の疑わしい予防接種後副反応の監視方案」を指す。